Large-scale semi-synthesis of indirubin from Strobilanthes cusia leaf extract and novel indirubin derivatives active against HT29 colon cancer cells
Main Article Content
Abstract
In this study, indirubin with a purity exceeding 97% was synthesized from Strobilanthes cusia leaf extract and isatin. Subsequently, this was used as the starting material for the synthesis of eight new indirubin-3’-oxime derivatives. These compounds, bearing substituents containing a bromine atom (3b, 3c), an azide group (4a–c), or 2-amino-1,3,4-thiadiazole moiety (5a–c), were successfully synthesized in yields ranging from 75% to 88%. The chemical structures of all compounds were elucidated using modern spectroscopic techniques, including 1H and 13C NMR spectroscopy, High-Resolution Electrospray Ionization Mass Spectrometry (HR-MS (ESI)), and Infra Red (IR) spectroscopy. The cytotoxic activity of the synthesized compounds was evaluated against the HT29 human colon cancer cell line. Indirubin derivatives 5b and 5c exhibited potent cytotoxic activity, with IC50 values of 3.2 ± 0.1 µM and 6.2 ± 0.2 µM, respectively. Both compounds were more potent than the parent indirubin (1a) and the positive control, 5-fluorouracil. Overall, the synthesized indirubin derivatives represent a valuable synthetic platform for the further development of novel indirubin-based bioactive compounds in medicinal chemistry and anticancer drug discovery.
Keywords
Indirubin, Oxime, Strobilanthes cusia, Synthetic modification, Anticancer activity, HT29 cell line
Article Details

This work is licensed under a Creative Commons Attribution 4.0 International License.
Introduction
Indirubin is an alkaloid with the molecular formula C16H10O2N2, contained in Indigo naturalis, including Strobilanthes cusia, which is widely distributed in Vietnam (Cuong et al., 2007, 2016). It is a well-known bioactive compound that acts as a potent inhibitor of Cyclin–Dependent Kinases (CDKs) through a competitive mechanism at the ATP-binding site (Hoessel et al., 1999), thereby interfering with key cellular processes and thereby suppressing cancer cell proliferation. In addition to its extensively studied anticancer activity, indirubin also exhibits a range of other important biological potencies, including antiviral (Mak et al., 2004), anti-inflammatory (Kunikata et al., 2000), and antifungal (Ponnusamy et al., 2010) activities.
Numerous studies have focused on the structural modification of indirubin derivatives with the aim of enhancing its antiproliferative activity against cancer cells (Lei et al., 2017; Nguyen et al., 2019, 2020; Nguyen & Le, 2024; Sogawa et al., 2025), increasing its aqueous solubility and consequently improving bioavailability (Cheng et al., 2017; Lei et al., 2017; Nguyen & Le, 2024), as well as identifying derivatives with antiplatelet aggregation (Lee et al., 2014) or neuroprotective effects (Sharma & Taliyan, 2014). These findings demonstrate that the design and synthesis of indirubin derivatives remain a promising and highly relevant strategy, attracting considerable interest from the scientific community. The development of novel indirubin derivatives bearing flexible and readily transformable functional groups is essential for advancing the design, synthesis, and structural expansion of indirubin-based derivatives.
This study reports the synthesis of novel 1-substituted indirubin-3’-oximes, bearing substituents containing a bromine atom, an azide group, or 2-amino-1,3,4-thiadiazole moiety (Figure 1). These functionalities are important in drug design: the amino group can be readily transformed into amide or azomethine derivatives; the azido group serves as a versatile precursor for click chemistry, enabling the construction of 1,2,3-triazole moiety; and the bromo substituent can participate in a variety of nucleophilic substitution reactions, facilitating the generation of structurally diverse derivatives. Collectively, these intermediates are expected to enable target-oriented synthesis of a wide range of novel indirubin derivatives, thereby contributing to the advancement of pharmaceutical chemistry and future drug discovery and development.
Figure 1. Synthesis of novel side-chain functionalized derivatives
Materials and Methods
Materials
Leaves and stem of Strobilanthes cusia (Nees) Kunte were collected from Ban Cay, Hoang Su Phi commune, Tuyen Quang Province, Vietnam in August 21, 2025 (22°43'54"N and 104°40'01"E). The plant material was taxonomically authenticated by Dr. Vu Dinh Duy at the Joint Vietnam–Russia Tropical Science and Technology Research Center. A voucher speciucher No. HSP05) was deposited at the Joint Vietnam–Russia Tropical Science and Technology Research Center.
All the reagents were of analytical grade with purities exceeding 97%. All solvents used for reactions, thin-layer chromatography (TLC) and column chromatography had purities greater than 99.5%. The reagents and solvents were purchased from reputable chemical suppliers and used as received without any further purification.
The 1H- and 13C-NMR spectra were measured by Bruker Avance 500 and Avance Neo 600 MHz spectrometers, using DMSO-d6 as solvent at the Institute of Chemistry, Vietnam Academy of Science and Technology; the chemical shifts δ were measured in ppm with respect to the solvent (1Н: δ = 2.50 ppm; 13C: δ = 39.50 ppm). The HR-MS(ESI) spectra were recorded on Xevo G2-XS QToF Mass spectrometer at the Institute for drug quality control Ho Chi Minh city. The IR spectra were obtained using PerkinElmer spectrometer; the melting point of compounds was determined using a Kofler hot stage. The nitrogen contents were evaluated by Kjeldahl method on the UDK 130D (Velp) system at the Joint Vietnam–Russia Tropical Science and Technology Research Center. TLC was used to monitor the reaction progress, select the solvent systems for column chromatography, and perform initial purity assessment of indirubin. Silica-gel (particle size 0.040–0.063 mm) was employed as the stationary phase for column chromatography.
Methods
Synthesis of crude indirubin from isatin and Strobilanthes cusia leaf extract
Crude indirubin was synthesized from Strobilanthes cusia leaf extract and isatin according to the procedure reported by Cuong et al. (2016) with minor modifications.
10 kg of fresh leaves were cleaned and then soaked in 30 L of water in an 80 L plastic container equipped with a tight-fitting lid. The mixture was allowed to ferment naturally for 5 days at 25-30°C. After fermentation, the extract was separated. The solid residue was squeezed and washed with an additional 5 L of water. The combined extracts were transferred to an 80 L heated stirring vessel, stirred at 200 rpm, and heated to 80°C. Subsequently, a solution of 90 g of isatin in 5 L of ethanol (96%) was added to the reaction mixture. The pH was adjusted to approx. 9-10 using an aq. Ca(OH)2 solution. The mixture was maintained at 80°C for 3 hours, then cooled to room temperature, and filtered through a Büchner funnel. The obtained solid was resuspended in 3 L of 1M HCl and stirred for 30 min, then filtered and washed thoroughly with distilled water until the filtrate reached pH 6–7. Finally, the solid collected on the funnel was dried at 105°C for 3 h, yielding 120 g of crude indirubin.
Evaluation of the purity of indirubin samples by thin layer chromatography
An indirubin sample (20 mg) was completely dissolved in 1.0 mL of DMF to obtain the original solution (S1). Then, a 100 µL aliquot of S1 was taken and diluted with 400 µL of acetone to obtain a working solution (S2). Subsequently, 10 µL of S2 was spotted onto a silica gel F254 TLC plate, and the solvents were allowed to evaporate at room temperature before analyzed by TLC using n-hexan/acetone (1.5:1, v/v) as the mobile phase.
Determination of total nitrogen content by the Kjeldahl method
The total nitrogen content of indirubin was determined by the Kjeldahl method according to the reported procedure (Abrams et al., 2014). In this experiment, 131 mg of indirubin was treated with a mixture of concentrated H2SO4 and K2SO4/CuSO4·5H2O as a catalyst at 400°C for 75 minutes to convert all nitrogen present in indirubin into ammonium ions (NH4+). After treatment, the mixture was cooled to room temperature and then alkalized with excess 35% NaOH solution to release gaseous ammonia (NH3). The gaseous ammonia was distilled off with steam and completely absorbed into 4% boric acid (H3BO3) solution. A few drops of Shiro–Tashiro indicator were then added to the obtained solution before titration with 0.1N HCl. The titration was completed when the color of the solution changed from green to reddish-purple. The number of moles of nitrogen in the indirubin sample was determined based on the number of moles of HCl consumed during titration. Based on this value, the total nitrogen content was calculated, as well as the indirubin content of the sample, based on the corresponding molecular formula.
Purification of crude indirubin by recrystallization
The recrystallization conditions of crude indirubin were investigated using the following procedure. Four portions of crude indirubin (2.5 g per sample) were placed in four two-necked round-bottom flasks equipped with a magnetic stirring bar, condenser, and thermometer. DMF (250 mL) was added to each flask, and the mixtures were heated with stirring at 120–130°C for 30 min until complete dissolution. Then the hot solution was filtered while hot to afford a clear filtrate. Different volumes of distilled water were added to the filtrates to adjust the DMF concentrations to 60%, 70%, 80%, and 90% (v/v), respectively. Each mixture was reheated to 100°C for 10 min, then allowed to cool slowly to room temperature and left undisturbed for 24 h to initiate crystallization. The resulting precipitate was collected by vacuum filtration through a Büchner funnel, washed with 70% ethanol (3 × 50 mL), and finally dried at 105°C for 3 h, yielding indirubin (1a).
Preparation of compounds 1b, 1c
The compounds 1b and 1c were synthesized according to the procedure described in the literature (Nguyen et al., 2020).
Preparation of compounds 2a, 2b, 2c
The compounds 2a, 2b and 2c were synthesized according to the procedure described in the literature (Nguyen & Le, 2024).
General procedure for the synthesis of indirubin derivatives (3a-c)
A round-bottom flask was charged with compound 2a–c (1.0 equiv, 1.25 mmol; 346 mg of 2a, 485 mg of 2b, or 488 mg of 2c), DMF (30 mL), triethylamine (4.0 equiv, 5.0 mmol, 0.70 mL), and 1,2-dibromoethane (4.0 equiv, 5.0 mmol, 0.44 mL). The reaction mixture was stirred at room temperature for 4 days. Then ethyl acetate (400 mL) was added, and the mixture was washed with 3% aq. Na2SO4 solution (3 × 400 mL). The organic layer was dried over anhydrous Na2SO4 for 12 h and filtered. The solvent was removed under reduced pressure to afford the crude product, which was purified by column chromatography (silica gel; n-hexane/acetone, 2:1, v/v).
General procedure for the synthesis of indirubin derivatives (4a-c)
A round-bottom flask was charged with compound 3a–c (1.0 equiv, 1.25 mmol; 480 mg of 3a, 619 mg of 3b, or 621 mg of 3c), DMSO (25 mL), and sodium azide (2.0 equiv, 2.5 mmol, 163 mg). The reaction mixture was stirred at 50°C for 10 h. Then ethyl acetate (500 mL) was added, and the mixture was washed with 3% aq. Na2SO4 solution (3 × 500 mL). The organic layer was dried over anhydrous Na2SO4 for 12 h and filtered. The solvent was removed under reduced pressure to afford the crude product, which was purified by column chromatography (silica gel; n-hexane/acetone, 2:1, v/v).
General procedure for the synthesis of indirubin derivatives (5a–c)
A round-bottom flask was charged with compound 5a–c (1.0 equiv, 1.0 mmol; 384 mg of 3a, 495 mg of 3b, or 497 mg of 3c), triethylamine (2.0 equiv, 2.0 mmol, 0.28 mL), and 5-amino-1,3,4-thiadiazole-2-thiol (2.0 equiv, 2.0 mmol, 266 mg). The reaction mixture was stirred at room temperature for 24 h. Then, cold water (100 mL) was added with vigorous stirring and the mixture was left to stand for 30 min. The resulting crude solid was filtered by vacuum filtration through a Büchner funnel and washed with distilled water (3 × 50 mL) and then with 70% ethanol (30 mL). The resulting solid was then dried under reduced pressure at 60°C for 5 h to afford compounds 5a–c.
Evaluation of cytotoxicity of indirubin derivatives
The inhibitory effect of compounds 1a and 5a–c on the proliferation of HT29 human colon cancer cells was evaluated using the MTT assay, as previously reported (Mosmann, 1983). In brief, HT-29 cells were kindly provided by Prof. Chi-Ying F Huang, National Yang Ming Chiao Tung University, Taipei, Taiwan. The cells were thawed and cultured in a T25 flask with Dulbecco's Modified Eagle medium containing 10% Fetal Bovine Serum and 1% Peniciline/Streptomycin. When grown to 70-80% confluence, HT29 were seeded into a 96-well plate at a cell density of 4 × 104 cells/mL and incubated overnight at 37°C and 5% CO2. Then, the cells were treated with compounds (10, 5, 2.5, 1.25, 0.625, 0.3125, 0.15625 µM). After 48h, MTT (1 mg/mL) was added to the plate to form formazan crystals, which were dissolved in 100% DMSO. The optical density was measured at 540nm using a microplate reader, and cell viability was calculated.
Results
Result of synthesis and evaluation of the purity of crude indirubin
From 10 kg of fresh leaves of Strobilanthes cusia, after the preparation with a solution of isatin (90 g) in ethanol 96% (5 L), 120 g of crude indirubin was obtained. The purity of crude indirubin was evaluated by TLC, and the result was showed on the Figure 2A.
Result of crude indirubin purification by recrystallization
The purification results are summarized in Table 1. Recrystallization afforded indirubin (1a) with a relative purity of 97.5 ± 0.3%, as determined by Kjeldahl analysis, confirmed by TLC (Figure 2B) and NMR spectroscopy.
Table 1. Results of indirubin purification by recrystallization method
Exp. No. | DMF concentration, (vol.%) | Indirubin weight, (g) | Presence of a blue spot of indigo on TLC after purification |
1 | 60 | 2.14 ± 0.05 | Yes |
2 | 70 | 1.97 ± 0.08 | Yes |
3 | 80 | 1.54 ± 0.04 | No |
4 | 90 | 0.98 ± 0.05 | No |
(Z)-[2,3'-biindolinylidene]-2',3-dione (indirubin) (1a)
Yield 60%, dark purple solid, m.p. 313-315°C; 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 11.00 (s, 1H), 10.87 (s, 1H), 8.76 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.62 – 7.52 (m, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.25 (td, J = 7.6, 1.2 Hz, 1H), 7.04 – 7.01 (m, 2H), 6.90 (d, J = 7.7 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 188.6, 170.9, 152.5, 140.9, 138.3, 137.1, 129.2, 124.7, 124.3, 121.4, 121.2, 121.2, 119.0, 113.4, 109.6, 106.6. HR-MS(ESI) calculated for C16H11N2O2: [M+H]+ (m/z): 263.0821, found: 263.0826
Result of compounds 1b, 1c preparation
Compounds 1b, 1c were obtained in 86% and 78% isolated yields, respectively.
(Z)-1'-(2-(piperidin-1-yl)ethyl)-[2,3'-biindolinylidene]-2',3-dione (1b)
Yield 86%, dark purple solid, m.p. 198-200°C; 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 10.93 (s, 1H, NH), 8.79 (d, J = 7.7 Hz, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.61 – 7.54 (m, 1H), 7.39 (dt, J = 8.1, 0.7 Hz, 1H), 7.32 (dt, J = 7.7, 1.2 Hz, 1H), 7.12 – 7.00 (m, 3H), 3.91 (t, J = 6.8 Hz, 2H), 2.58 (t, J = 6.8 Hz, 2H), 2.48 – 2.40 (m, 4H), 1.51 – 1.43 (m, 4H), 1.41 – 1.33 (m, 2H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 188.0, 168.9, 152.1, 141.2, 138.3, 136.7, 128.8, 124.2, 124.0, 121.3, 121.0, 120.5, 118.9, 113.0, 108.3, 105.5, 55.3, 53.7, 37.1, 25.2, 23.6. HR-MS(ESI) calculated for C23H24N3O2: [M+H]+ (m/z): 374.1869, found: 374.1864.
(Z)-1'-(2-morpholinoethyl)-[2,3'-biindolinylidene]-2',3-dione (1c)
Yield 78%, dark purple solid, m.p. 195-197°C; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 11.06 (s, 1H, NH), 8.80 (d, J = 7.7 Hz, 1H), 7.66 (d, J = 7.4 Hz, 1H), 7.61 – 7.55 (m, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.13 (d, J = 7.8 Hz, 1H), 7.08 (t, J = 7.6 Hz, 1H), 7.03 (t, J = 7.4 Hz, 1H), 3.94 (t, J = 6.6 Hz, 2H), 3.57 – 3.47 (m, 4H), 2.59 (t, J = 6.6 Hz, 2H), 2.48 – 2.44 (m, 4H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 188.5, 169.1, 152.4, 141.2, 138.6, 137.2, 129.1, 124.5, 124.4, 121.7, 121.4, 120.7, 119.0, 113.4, 108.6, 105.5, 66.2, 55.3, 53.2, 36.6. HR-MS(ESI) calculated for C22H22N3O3: [M+H]+ (m/z): 376.1661, found: 376.1657.
Result of compounds 2a-c preparation
Compounds 2a–c were obtained in 68–74% isolated yields.
(2Z,3E)-3-(hydroxyimino)-[2,3'-biindolinylidene]-2'-one (2a)
Yield 73%, bright red solid, m.p. 247-248°C; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 13.44 (s, 1H, OH), 11.74 (s, 1H, NH), 10.69 (s, 1H, NH), 8.66 (d, J = 7.8 Hz, 1H), 8.24 (d, J = 7.6 Hz, 1H), 7.42 – 7.37 (m, 2H), 7.13 (t, J = 7.5 Hz, 1H), 7.03 – 7.00 (m, 1H), 6.96 (t, J = 7.6 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 170.9, 151.3, 145.2, 144.8, 138.3, 131.9, 127.9, 125.8, 122.9, 122.6, 121.4, 120.3, 116.5, 111.4, 108.8, 98.9. HR-MS(ESI) calculated for C16H12N3O2: [M+H]+ (m/z): 278.0930, found: 278.0923.
(2Z,3E)-3-(hydroxyimino)-1'-(2-(piperidin-1-yl)ethyl)-[2,3'-biindolinylidene]-2'-one (2b)
Yield 74%, bright red solid, m.p. 233-235°C; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 13.80 – 12.80 (br s, 1H, OH), 11.75 (s, 1H, NH), 8.70 (dd, J = 7.8, 1.2 Hz, 1H), 8.24 (d, J = 7.7 Hz, 1H), 7.42 – 7.38 (m, 2H), 7.20 (td, J = 7.6, 1.3 Hz, 1H), 7.07 (d, J = 7.7 Hz, 1H), 7.05 – 7.01 (m, 1H), 7.01 – 6.98 (m, 1H), 3.95 (t, J = 7.0 Hz, 2H), 2.54 (t, J = 7.0 Hz, 2H), 2.48 – 2.39 (m, 4H), 1.46 (quintet, J = 5.6 Hz, 4H), 1.38 – 1.33 (m, 2H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 168.9, 151.3, 145.5, 144.7, 138.8, 132.0, 127.9, 125.8, 122.8, 121.9, 121.5, 120.7, 116.5, 111.5, 107.8, 97.9, 55.9, 54.1, 36.9, 25.5, 23.9. HR-MS(ESI) calculated for C23H25N4O2: [M+H]+ (m/z): 389.1978, found: 389.1973.
(2Z,3E)-3-(hydroxyimino)-1'-(2-morpholinoethyl)-[2,3'-biindolinylidene]-2'-one (2c)
Yield 68%, bright red solid, m.p. 237-238°C; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 13.52 (s, 1H, OH), 11.74 (s, 1H, NH), 8.70 (d, J = 7.8 Hz, 1H), 8.24 (d, J = 7.6 Hz, 1H), 7.39 (t, J = 3.9 Hz, 2H), 7.20 (td, J = 7.7, 0.8 Hz, 1H), 7.09 (d, J = 7.7 Hz, 1H), 7.05 – 6.98 (m, 2H), 3.97 (t, J = 6.8 Hz, 2H), 3.57 – 3.50 (m, 4H), 2.59 (t, J = 6.8 Hz, 2H), 2.50 – 2.44 (m, 4H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 169.0, 151.3, 145.5, 144.7, 138.8, 132.0, 127.9, 125.8, 122.8, 121.9, 121.6, 120.8, 116.5, 111.5, 107.8, 97.9, 66.2, 55.6, 53.3, 36.5. HR-MS(ESI) calculated for C22H23N4O3: [M+H]+ (m/z): 391.1770, found: 391.1767.
Result of compounds 3a-c preparation
Compounds 3a–c were obtained in 75–80% isolated yields.
(2Z,3E)-3-((2-bromoethoxy)imino)-[2,3'-biindolinylidene]-2'-one (3a)
Yield 80%, bright red solid, m.p. 221-223°C; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 11.67 (s, 1H, NH), 10.75 (s, 1H, NH), 8.56 (d, J = 7.8 Hz, 1H), 8.22 (d, J = 7.7 Hz, 1H), 7.47 – 7.42 (m, 1H), 7.41 (d, J = 7.9 Hz, 1H), 7.16 (td, J = 7.6, 0.8 Hz, 1H), 7.06 – 7.01 (m, 1H), 7.01 – 6.96 (m, 1H), 6.91 (d, J = 7.6 Hz, 1H), 4.91 (t, J = 5.4 Hz, 2H), 3.98 (t, J = 5.4 Hz, 2H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 170.8, 152.0, 145.6, 143.5, 138.7, 133.0 128.6, 126.5, 123.2, 122.1, 121.3, 120.6, 116.0, 111.7, 108.9, 100.6, 75.8, 31.1. HR-MS(ESI) calculated for C18H15N3O2Br: [M+H]+ (m/z): 384.0348; 386.0327, found: 384.0345; 386.0327.
(2Z,3E)-3-((2-bromoethoxy)imino)-1'-(2-(piperidin-1-yl)ethyl)-[2,3'-biindolinylidene]-2'-one (3b)
Yield 75%, bright red solid, m.p. 161-162°C; 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.71 (s, 1H, NH), 8.61 (d, J = 7.7 Hz, 1H), 8.21 (d, J = 7.6 Hz, 1H), 7.48 – 7.40 (m, 2H), 7.23 (td, J = 7.7, 0.8 Hz, 1H), 7.09 (d, J = 7.7 Hz, 1H), 7.06 – 7.01 (m, 2H), 4.91 (t, J = 5.6 Hz, 2H), 3.98 (t, J = 5.6 Hz, 2H), 3.94 (t, J = 6.9 Hz, 2H), 2.54 (t, J = 6.9 Hz, 2H), 2.47 – 2.40 (m, 4H), 1.49 – 1.43 (m, 4H), 1.39 – 1.33 (m, 2H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm):168.7, 151.9, 145.5, 143.7, 139.3, 133.0, 128.5, 126.4, 123.0, 121.5, 121.4, 121.0, 116.0, 111.8, 107.9, 99.6, 75.8, 55.8, 54.1, 36.9, 31.1, 25.5, 23.9. HR-MS(ESI) calculated for C25H28N4O2Br: [M+H]+ (m/z): 495.1396; 497.1375, found: 495.1395; 497.1378.
(2Z,3E)-3-((2-bromoethoxy)imino)-1'-(2-morpholinoethyl)-[2,3'-biindolinylidene]-2'-one (3c)
Yield 77%, bright red solid, m.p. 191-193°C; 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.63 (s, 1H, NH), 8.62 (d, J = 7.8 Hz, 1H), 8.23 (d, J = 7.6 Hz, 1H), 7.45 (td, J = 8.0, 1.2 Hz, 1H), 7.37 (d, J = 7.9 Hz, 1H), 7.24 (td, J = 7.7, 1.1 Hz, 1H), 7.09 (d, J = 7.8 Hz, 1H), 7.07 – 7.02 (m, 2H), 4.97 – 4.86 (m, 2H), 4.04 – 3.91 (m, 4H), 3.61 – 3.51 (m, 4H), 2.64 (t, J = 6.8 Hz, 2H), 2.53 – 2.50 (m, 4H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 168.8, 151.7, 145.2, 143.6, 139.0, 132.7, 128.3, 126.1, 122.8, 121.3, 121.2, 120.7, 115.9, 111.3, 107.6, 99.5, 75.6, 65.9, 55.2, 52.9, 36.5, 30.4. HR-MS(ESI) calculated for C24H26N4O3Br: [M+H]+ (m/z): 497.1188; 499.1168, found: 497.1189; 499.1172.
Result of compounds 4a-c preparation
Compounds 4a–c were obtained in 77–85% isolated yields.
(2Z,3E)-3-((2-azidoethoxy)imino)-[2,3'-biindolinylidene]-2'-one (4a)
Yield 85%, bright red solid, m.p. 205-207°C ; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 11.67 (s, 1H, NH), 10.75 (s, 1H, NH), 8.59 (d, J = 7.8 Hz, 1H), 8.18 (d, J = 7.7 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.39 (d, J = 7.9 Hz, 1H), 7.16 (t, J = 7.6 Hz, 1H), 7.04 – 6.96 (m, 2H), 6.91 (d, J = 7.6 Hz, 1H), 4.81 – 4.73 (m, 2H), 3.88 – 3.80 (m, 2H).13C NMR (DMSO-d6, 150 MHz) δ (ppm): 170.9, 151.9, 145.6, 143.6, 138.8, 133.0, 128.5, 126.5, 123.3, 122.2, 121.4, 120.7, 116.1, 111.7, 109.0, 100.6, 75.1, 49.8. HR-MS(ESI) calculated for C18H15N6O2: [M+H]+ (m/z): 347.1256, found: 347.1252.
(2Z,3E)-3-((2-azidoethoxy)imino)-1'-(2-(piperidin-1-yl)ethyl)-[2,3'-biindolinylidene]-2'-one (4b)
Yield 77%, bright red solid, m.p. 118-120°C; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 11.72 (s, 1H, NH), 8.64 (d, J = 7.9 Hz, 1H), 8.19 (d, J = 7.6 Hz, 1H), 7.47 – 7.40 (m, 2H), 7.24 (td, J = 7.6, 1.1 Hz, 1H), 7.10 (d, J = 7.7 Hz, 1H), 7.07 – 7.02 (m, 2H), 4.82 – 4.75 (m, 2H), 3.95 (t, J = 6.9 Hz, 2H), 3.89 – 3.83 (m, 2H), 2.54 (t, J = 6.9 Hz, 2H), 2.47 – 2.39 (m, 4H), 1.49 – 1.42 (m, 4H), 1.39 – 1.33 (m, 2H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 168.8, 151.8, 145.5, 143.8, 139.3, 133.0, 128.4, 126.4, 123.1, 121.4 (2C), 121.0, 116.0, 111.8, 107.9, 99.5, 75.1, 55.8, 54.1, 49.8, 36.9, 25.5, 23.9. HR-MS(ESI) calculated for C25H28N7O2: [M+H]+ (m/z): 458.2304, found: 458.2302.
(2Z,3E)-3-((2-azidoethoxy)imino)-1'-(2-morpholinoethyl)-[2,3'-biindolinylidene]-2'-one (4c)
Yield 80%, bright red solid, m.p. 144-146°C; 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 11.71 (s, 1H, NH), 8.65 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 7.7 Hz, 1H), 7.47 – 7.40 (m, 2H), 7.24 (t, J = 7.6 Hz, 1H), 7.12 (d, J = 7.8 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.3 Hz, 1H), 4.80 – 4.75 (m, 2H), 3.98 (t, J = 6.8 Hz, 2H), 3.88 – 3.82 (m, 2H), 3.57 – 3.50 (m, 4H), 2.59 (t, J = 6.8 Hz, 2H), 2.49 – 2.45 (m, 4H).13C NMR (DMSO-d6, 150 MHz) δ (ppm): 168.9, 151.8, 145.5, 143.8, 139.2, 133.0, 128.4, 126.4, 123.1, 121.5 (2C), 121.0, 116.0, 111.8, 108.0, 99.5, 75.1, 66.1, 55.5, 53.2, 49.8, 36.5. HR-MS(ESI) calculated for C24H26N7O3: [M+H]+ (m/z): 460.2097, found: 460.2096.
Result of compounds 5a-c preparation
Compounds 5a–c were obtained in 82–88% isolated yields.
(2Z,3E)-3-((2-((5-amino-1,3,4-thiadiazol-2-yl)thio)ethoxy)imino)-[2,3'-biindolinylidene]-2'-one (5a)
Yield 88%, dark red solid, m.p. 219-221°C; 1H NMR (600 MHz, DMSO-d6) δ (ppm): δ 11.67 (s, 1H, NH), 10.73 (s, 1H, NH), 8.55 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 7.7 Hz, 1H), 7.45 – 7.41 (m, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.29 (s, 2H), 7.14 (td, J = 7.6, 0.8 Hz, 1H), 7.03 – 6.99 (m, 2H), 6.90 (d, J = 7.6 Hz, 1H), 4.84 (t, J = 6.3 Hz, 2H), 3.65 (t, J = 6.3 Hz, 2H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 170.8, 169.6, 151.8, 149.2, 145.5, 143.6, 138.7, 132.9, 128.5, 126.4, 123.3, 122.2, 121.3, 120.7, 116.0, 111.6, 108.9, 100.4, 74.5, 33.1. HR-MS(ESI) calculated for C20H17N6O2S2: [M+H]+ (m/z): 437.0854, found: 437.0856.
(2Z,3E)-3-((2-((5-amino-1,3,4-thiadiazol-2-yl)thio)ethoxy)imino)-1'-(2-(piperidin-1-yl)ethyl)-[2,3'-biindolinylidene]-2'-one (5b)
Yield 82%, dark red solid, m.p. 202-205°C; 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.70 (s, 1H, NH), 8.60 (d, J = 7.7 Hz, 1H), 8.11 (d, J = 7.7 Hz, 1H), 7.45 – 7.42 (m, 1H), 7.40 (d, J = 7.8 Hz, 1H), 7.29 (s, 2H), 7.23 – 7.19 (m, 1H), 7.09 – 7.04 (m, 2H), 7.04 – 7.00 (m, 1H), 4.84 (t, J = 6.3 Hz, 2H), 3.93 (t, J = 6.9 Hz, 2H), 3.65 (t, J = 6.3 Hz, 2H), 2.53 (t, J = 7.0 Hz, 2H), 2.48 – 2.39 (m, 4H), 1.48-1.44 (m, 4H), 1.39 – 1.32 (m, 2H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 169.61, 168.76, 151.72, 149.16, 145.43, 143.77, 139.17, 132.89, 128.48, 126.29, 123.10, 121.43, 121.39, 121.09, 116.03, 111.67, 107.84, 99.47, 74.51, 55.83, 54.03, 36.90, 33.07, 25.49, 23.86. HR-MS(ESI) calculated for C27H30N7O2S2: [M+H]+ (m/z): 548.1902, found: 548.1903.
(2Z,3E)-3-((2-((5-amino-1,3,4-thiadiazol-2-yl)thio)ethoxy)imino)-1'-(2-morpholinoethyl)-[2,3'-biindolinylidene]-2'-one (5c)
Yield 84%, dark red solid, m.p. 218-220°C; 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.70 (s, 1H, NH), 8.61 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 7.7 Hz, 1H), 7.47 – 7.39 (m, 2H), 7.29 (s, 2H), 7.22 (t, J = 7.6 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 7.07 (t, J = 7.6 Hz, 1H), 7.03 (t, J = 7.4 Hz, 1H), 4.85 (t, J = 6.3 Hz, 2H), 3.97 (t, J = 6.7 Hz, 2H), 3.66 (t, J = 6.3 Hz, 2H), 3.58 – 3.49 (m, 4H), 2.59 (t, J = 6.8 Hz, 2H), 2.49 – 2.44 (m, 4H). 13C NMR (DMSO-d6, 150 MHz) δ (ppm): 169.6, 168.8, 151.7, 149.2, 145.4, 143.8, 139.1, 132.9, 128.5, 126.3, 123.1, 121.5, 121.4, 121.2, 116.0, 111.7, 107.9, 99.4, 74.5, 66.1, 55.5, 53.2, 36.5, 33.1. HR-MS(ESI) calculated for C26H28N7O3S2: [M+H]+ (m/z): 550.1695, found: 550.1699.
Results of cytotoxicity evaluation of indirubin derivatives
The inhibitory effect of compounds 1a and 5a–c on the proliferation of HT29 human colon cancer cells has been presented in Table 2.
Table 2. Cytotoxicity of 1a and 5a–c against HT29 colon cancer cell line
Exp. No. | Compound | IC50 (µM) |
1 | 1a | >10 |
2 | 5a | >10 |
3 | 5b | 3.2 ± 0.1 |
4 | 5c | 6.2 ± 0.2 |
5 | 5-Fluorouracil | >10 |
Discussion
Chemistry
The purity of the crude indirubin was preliminarily evaluated by TLC using n-hexane/acetone (1.5:1, v/v) as the eluent.
In the Figure 2A, the chromatogram shows a main purple spot with a Rf value of 0.58 and a blue impurity spot with a Rf value of 0.65, which is attributed to indigo. To obtain higher purity indirubin suitable for the synthesis of indirubin-3’-oximes, the blue-colored impurity present in the crude indirubin was removed by recrystallization.
A |
B |
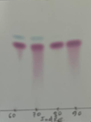
Figure 2. TLC plots of crude indirubin (A) and indirubin after purification (B) using the DMF-H2O solvent system
Applying the recrystallization procedure described in Section 2.2.4, the purification results (Table 1) indicate that the indigo impurity (appeared as a blue spot on TLC plate) remained in recrystallized indirubin obtained in the experiments 1 and 2 (corresponding to the solvent system containing 60% and 70% DMF, respectively). When the concentration of DMF was increased to 80 or 90%, no detectable impurity was observed on TLC (Figure 2B). However, the isolated yield of pure indirubin obtained using 80% DMF (1.54 ± 0.04 g) was significantly higher than that obtained with 90% DMF (0.98 ± 0.05 g). Therefore, the optimal condition for recrystallization of crude indirubin was identified as a DMF–water solvent system with a ratio of 80:20 (v/v), which was subsequently applied according to the procedure described in Section 2.2.4.
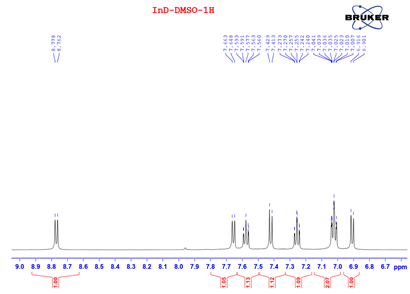
Figure 3. 1H NMR spectrum of indirubin 1a
Figure 4. 13C NMR spectrum of indirubin 1a
The structure of indirubin was confirmed by 1H- and 13C-NMR spectroscopy and high-resolution mass spectrometry (HR-MS (ESI). The 1H and 13C NMR spectra of indirubin are shown in Figures 3 and 4, respectively. All observed signals in NMR spectra were in full agreement with the reported data for indirubin [1]. Furthermore, the HR-MS (ESI) analysis showed a prominent ion peak at m/z 263.0826 [M+H]+, corresponding to the molecular formula C16H11N2O2, confirming the identity of the compound as indirubin.
Recrystallization afforded indirubin (1a) in high yield with a relative purity of 97.5 ± 0.3%, as determined by Kjeldahl analysis confirmed by TLC and NMR spectroscopy. This efficient purification protocol yields material of sufficient quality for direct use as a starting material in subsequent synthetic steps and represents a significant improvement over previously reported methods. The corresponding functionalized indirubin-3’-oximes were synthesized from indirubin (1a) through a series of chemical transformations, as shown in Scheme 1.
Reagents and conditions: i) 1-(2-chloroethyl)piperidine hydrochloride or 1-(2-chloroethyl)morpholine hydrochloride/K2CO3, KI, TBAB, DMF, 65°C, 20 h, ii) NH2OH·HCl, K2CO3, EtOH/H2O, 78°C, 6 h, iii) 1,2-dibromoethane, TEA, DMF, rt, 4 days, iv) NaN3, DMSO, 50°C, 10 h, v) 5-amino-1,3,4-thiadiazole-2-thiol, TEA, DMF, rt, 24h.
Scheme 1. Synthesis of indirubin-3’-oximes
3a |
3b |
3c |
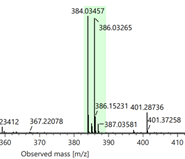
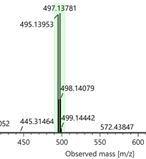
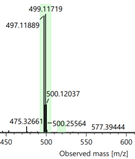
Figure 5. Part of the HR-MS(ESI) spectra of compounds 3a, 3b and 3c
4a |
4b |
4c |
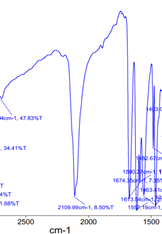


Figure 6. A part of IR spectra of compounds 4a, 4b and 4c
Compounds 1b–c and 2a–c were synthesized following a previously reported procedure (Nguyen & Le, 2024). The isolated yields for compounds 1b and 1c were 78% and 86%, respectively, while those for compounds 2a–c ranged from 68 to 74%. The structures of all compounds were unambiguously confirmed by 1H and 13C-NMR spectroscopy and HR-MS (ESI) analysis. O-Alkylation of oxime derivatives 2a–c with 1,2-dibromoethane, using triethylamine as a base in DMF solvent at room temperature for 4 days, afforded the corresponding bromo-containing derivatives 3a–c in high isolated yields (75–80%). Compounds 3b and 3c have not been previously reported. Their chemical structures were confirmed by 1H NMR, 13C NMR, and HR-MS (ESI) analyses. The HR-MS (ESI) spectra showed the expected isotopic pattern for a bromine atom, with two characteristic [M + H]+ ion peaks separated by 2 m/z units (Figure 5), corresponding to the natural abundance of 79Br and 81Br.
In the next step, bromo-containing derivatives of indirubin 3a–c were introduced into the reaction of nucleophilic substitution of the bromine atom under the action of sodium azide to form the corresponding derivatives 4a–c in 77–85% yield. Compounds 4a–c represent novel indirubin derivatives that have not been previously reported. Their chemical structures were unambiguously confirmed using 1H, 13C-NMR, HR-MS (ESI), and IR spectroscopy. The IR spectra of these compounds exhibit a strong absorption band in the range of 2101–2109 cm-1, which is characteristic of the azido group stretching vibration (Figure 6).
To further diversify the indirubin scaffold, compounds 3a–c were reacted with 5-amino-1,3,4-thiadiazole-2-thiol in DMF using triethylamine as a base. Following the work-up and purification procedure described in Section 2.2.7, the target compounds 5a–c were obtained in good isolated yields of 82–88%. Their chemical structures were unambiguously confirmed using 1H, 13C-NMR, HR-MS (ESI), and IR spectroscopy (Figure 7). In the 1H NMR spectra of derivatives 5a–c, characteristic signals corresponding to the –NH2 group were observed at approximately δ 7.29 ppm. In the IR spectra, two absorption bands were observed in the range of 3200–3400 cm-1, which are diagnostic of N–H stretching vibrations of the amino group.
A |
B |
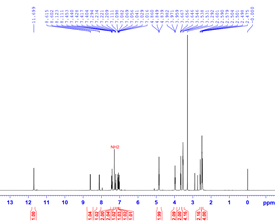
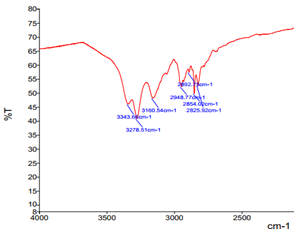
Figure 7. Part of the 1H NMR (A) and IR (B) of compound 5c
Evaluation of cytotoxic activity
Compounds 1a and 5a–c were tested against HT29 colon cancer cell line by MTT method to evaluate their anticancer activity. 5-Fluorouracil (FLU) was used as positive control. The results have been presented in Table 2 and Figure 8.
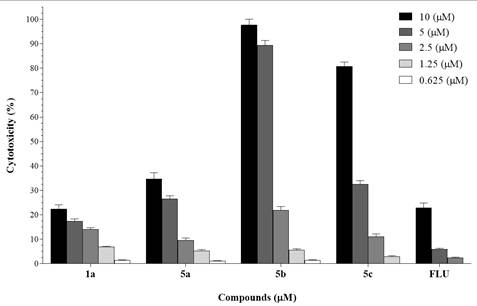
Figure 8. Dose-dependent responses of 1a and 5a–c towards HT29 cell proliferation
Compounds 1a and 5a did not exhibit significant cytotoxicity against HT29 cells (IC50 > 10 µM). In contrast, compounds 5b and 5c, containing piperidine and morpholine moieties, respectively, demonstrated significant cytotoxicity against the HT29 colon cancer cell line with IC50 values of 3.2 ± 0.1 and 6.2 ± 0.2 µM, respectively. Notably, the activities of these two compounds were superior to that of the positive control, 5-fluorouracil. These results suggest that modification of the indirubin structure is a potential approach that opens the possibility of developing new derivatives with promising pharmacological activity in the field of medicinal chemistry.
Conclusion
Indirubin 1a was synthesized from Strobilanthes cusia leaf extract and isatin in high yield and greater than 97% relative purity. Eight novel indirubin-3’-oximes were synthesized, including two brominated (3b, 3c), three azido-functionalized (4a–c), and three 2-amino-1,3,4-thiadiazole-containing derivatives (5a–c) in yields ranging from 75 to 88%. All structures were confirmed by 1H-NMR, 13C-NMR, HR-MS (ESI) and IR spectroscopy. Indirubin derivatives,5b and 5c, exhibited high cytotoxicity against HT29 colon cancer cells, with IC50 values of 3.2 µM and 6.2 µM, respectively. These results demonstrated that structural modification of the indirubin scaffold is a promising stratagy for developing new pharmacologically active compounds.
Acknowledgements: This work was funded by the project No. M-4.2 and the Ministry of Science and Higher Education of the Russian Federation (FFZZ-2025-0010) as part of work with the Joint Vietnam‒Russia Tropical Science and Technology Research Center (project M-4.2).
Data Availability Statement: The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Author contributions: Nguyen Trong Dan designed the study; Pham Duy Nam, Vu Dinh Duy, Vu Duy Nhan, Vo Thi Hoai Thu, Hoang Quang Cuong, and Hoang Duc Quang carried out the experiments and prepared the bioactivity evaluation; Dmitri I. Fomenkov, Alexey I. Ilovaisky, Yvan A. Yaremenko, Alexander O. Terent’ev, Nguyen Trong Dan, and Bui Thi Thu Trang analyzed the experimental data and wrote the manuscript.
Conflicts of interest statement: The authors declare no conflict of interest.
Statement on the use of Generative AI: The authors affirm that no generative AI tools were used to create or modify the scientific content of this manuscript. All analyses, interpretations, and conclusions are entirely the work of the authors.
References
Abrams, D., Metcalf, D., & Hojjatie, M. (2014). Determination of Kjeldahl nitrogen in fertilizers by AOAC Official Method 978.02: Effect of copper sulfate as a catalyst. Journal of AOAC International, 97(3), 764–767. https://doi.org/10.5740/jaoacint.13-299
Cheng, X., Merz, K.-H., Vatter, S., Zeller, J., Muehlbeyer, S., Thommet, A., Christ, J., Wölfl, S., & Eisenbrand, G. (2017). Identification of a water-soluble indirubin derivative as potent inhibitor of insulin-like growth factor 1 receptor through structural modification of the parent natural molecule. Journal of Medicinal Chemistry, 60(12), 4949–4962. https://doi.org/10.1021/acs.jmedchem.7b00324
Cuong, N. M., Khanh, P. N., Ha, V. T., Huong, T. T., Tung, N. T., Cuc, N. T., & Thao, D. T. (2016). Semi-synthesis of indirubin-3′-oxime from Strobilanthes cusia leaves, its acute and sub-chronic toxicity, in vitro and in vivo antitumor activity in Lewis lung carcinoma bearing mice. Journal of Pharmacognosy and Phytochemistry, 5, 292–301.
Cuong, N. M., Thuy, D. T. T., Ha, N. V., & Tai, B. H. (2007). Isolation of indirubin from the leaves of Strobilanthes cusia. Journal of Science and Technology, 45, 195–199.
Hoessel, R., Leclerc, S., Endicott, J. A., Noble, M. E. M., Lawrie, A., Tunnah, P., Leost, M., Damiens, E., Marie, D., Marko, D., Niederberger, E., Tang, W., Eisenbrand, G., & Meijer, L. (1999). Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nature Cell Biology, 1(1), 60–67. https://doi.org/10.1038/9035
Kunikata, T., Tatefuji, T., Aga, H., Iwaki, K., Ikeda, M., & Kurimoto, M. (2000). Indirubin inhibits inflammatory reactions in delayed-type hypersensitivity. European Journal of Pharmacology, 410(1), 93–100. https://doi.org/10.1016/S0014-2999(00)00879-7
Lee, J.-J., Han, J.-H., Jung, S.-H., Lee, S.-G., Kim, I.-S., Nguyen, M. C., Tran, T. H., Pham, N. K., Kim, Y. H., Yun, Y.-P., Ma, J. Y., & Myung, C.-S. (2014). Antiplatelet action of indirubin-3′-monoxime through suppression of glycoprotein VI-mediated signal transduction: A possible role for ERK signaling in platelets. Vascular Pharmacology, 63(3), 182–192. https://doi.org/10.1016/j.vph.2014.10.005
Lei, Z., Chengyue, Z., Yang, W., Jing, W., & Qizheng, Y. (2017). Synthesis of novel indirubin derivatives and their effects on the proliferation, cell cycle and apoptosis in acute myeloblastic leukemia HL-60 cells. Chinese Journal of Organic Chemistry, 37(6), 1523–1529. https://doi.org/10.6023/cjoc201704018
Mak, N.-K., Leung, C.-Y., Wei, X.-Y., Shen, X.-L., Wong, R. N.-S., Leung, K.-N., & Fung, M.-C. (2004). Inhibition of RANTES expression by indirubin in influenza virus-infected human bronchial epithelial cells. Biochemical Pharmacology, 67(1), 167–174. https://doi.org/10.1016/j.bcp.2003.08.020
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. Journal of Immunological Methods, 65(1–2), 55–63. https://doi.org/10.1016/0022-1759(83)90303-4
Nguyen, D. T., Truong, G. N., Vuong, T. V., Nguyen, T. N., Nguyen, M. C., To, D. C., Dinh Thi Thuy, T., Luu, V. C., & Tran, K. V. (2019). Synthesis of new indirubin derivatives and their in vitro anticancer activity. Chemical Papers, 73(5), 1083–1092. https://doi.org/10.1007/s11696-018-0659-4
Nguyen, T. D., & Le, T. M. D. (2024). Development of a novel indirubin derivative with enhanced anticancer properties: Synthesis, in vitro, and in vivo evaluation. Chemical Papers, 78(4), 2469–2478. https://doi.org/10.1007/s11696-023-03253-w
Nguyen, T. D., Hoang, D. Q., Vuong, V. T., Do, H. N., Nguyen, M. C., To, D. C., Tran, Q. T., Bach, G. L., Nguyen, H. T. A., Nguyen, T. M., Ngo, T. L., Luu, V. C., & Pham, M. Q. (2020). Design, synthesis, structure, in vitro cytotoxic activity evaluation and docking studies on target enzyme GSK-3β of new indirubin-3′-oxime derivatives. Scientific Reports, 10(1), 11429. https://doi.org/10.1038/s41598-020-68134-8
Ponnusamy, K., Petchiammal, C., Mohankumar, R., & Hopper, W. (2010). In vitro antifungal activity of indirubin isolated from a South Indian ethnomedicinal plant Wrightia tinctoria R. Br. Journal of Ethnopharmacology, 132(1), 349–354. https://doi.org/10.1016/j.jep.2010.07.050
Sharma, S., & Taliyan, R. (2014). Neuroprotective role of indirubin-3′-monoxime, a GSK-3β inhibitor in high fat diet induced cognitive impairment in mice. Biochemical and Biophysical Research Communications, 452(4), 1009–1015. https://doi.org/10.1016/j.bbrc.2014.09.034
Sogawa, K., Kato, K., Sano, M., Nakayoshi, T., Yoshioka, H., Kato, H., Oda, A., Funada, M., Suzuki, T., Kurihara, M., & Ichimaru, Y. (2025). Indirubin derivatives bearing an oxirane moiety are promising chemosensitizers for combination treatment in pancreatic cancer. Medicinal Chemistry Research. https://doi.org/10.1007/s00044-025-03499-x