Synthesis and in vitro cytotoxicity evaluation of murrayafoline a derived β-thioether alcohols
Main Article Content
Abstract
Three new β-thioether alcohol derivatives 7a-c of murrayafoline A with 1,3,4-oxadiazole-2-thiol derivatives 6a-c were successfully synthesized from murrayafoline A 1 and The corresponding thiols 6a-c by a simple three-step procedure involving N-alkylation of murrayafoline A with epichlorohydrin using NaH in THF yielding the intermediate 2, followed by N-glycydyl ring opening of 2 with 6a-c, which were prepared from the corresponding acids though steps including esterification with ethanol catalyzed by concentrated sulfuric acid, followed by nucleophilic substitution reaction in the refluxing point of methanol with hydrazine, and their cyclization of the obtained hydrazides with CS2 in potassium-ethanol to give 6a-c as the nucleophilic agents for the opening reaction of intermediate 2 yielding the designed targets 7a-c. The obtained target compounds were confirmed by spectroscopic data, including NMR and HRMS. Spectral data were in excellent agreement with their structures. Cytotoxic activity against A549 and Hep-2C cell lines was selected to explore the anticancer potencies of β-thioether alcohols 7a-c in comparison with the starting compound, murrayafoline A, and the positive control, ellipticine. The obtained results of chemistry and anticancer effects provided a useful profile for new structures of β-thioether alcohol derivatives 7a-c, as well as their biological activity toward A549 and Hep-2C cells. In addition, the safety evaluation of three β-thioether alcohols was conducted using normal cells VERO from the kidney of African monkeys. The results showed that they were non-toxic toward VERO cells. Among three β-thioether alcohols, only derivative 7a, containing a 4-methoxybenzyl group in the thiol moiety, exhibited weak cytotoxic activity against A549 and Hep-2C cell lines at the 50% inhibitory concentrations of 35.84 and 33.20 µM, respectively.
Keywords
Murrayafoline A derivatives, thioethers, conjugate, cytotoxicity, murrayafoline A, Synthesis
Article Details

This work is licensed under a Creative Commons Attribution 4.0 International License.
- Highlights:
Three new β-thioether alcohol derivatives 7a-c of murrayafoline A with 1,3,4-oxadiazole-2-thiol derivatives were prepared and structurally elucidated.
The derivative 7a, containing a 4-methoxybenzyl group in the thiol moiety, expressed weak cytotoxic activity against human cell lines: A549 and Hep-2C 50% at the inhibitory values of 35.84 and 33.20 µM, respectively.
1. INTRODUCTION
Natural heterocyclic compounds containing nitrogen have played an important role in medicinal chemistry. They are present in various classes under different structures, including alkaloids, nucleobases, nucleosides, amino acids, amines, and others [1-3]. Among natural nitrogen-containing compounds, indole-containing compounds can be found in many plant kingdoms, which possess various bioactivities, including anticancer, antibacterial, and anti-inflammatory effects [4,5]. Several natural alkaloids are drugs or potent candidates for the research and development of new medications. For example, the berberin from Coscinium fenestratum is used to treat digestive disorders [6]. The alkaloid vinblastine from Catharanthus roseus and its derivatives, as well as camptothecin from Camptotheca acuminata, are used for cancer treatment [7,8] (Figure 1).
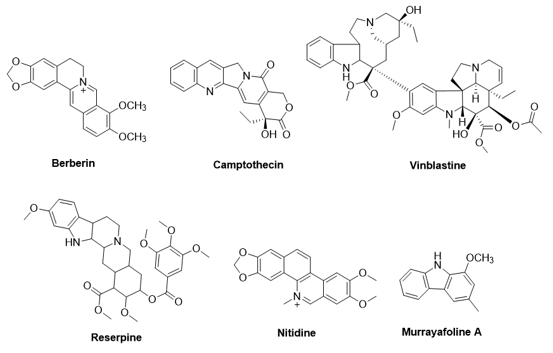
Figure 1: Some bioactive natural alkaloids
In Vietnam, in addition to the known alkaloids mentioned above, several other natural alkaloids have also been considered as potent bioactive compounds for research and drug development, such as reserpine from Rauvolfia verticillata [9], Nitidine from the bark of Zanthoxylum myriacanthum [10], and murrayafoline A from Glycosmis stenocarpa [11] (Figure 1). Notably, murrayafoline A, a bioactive bis-indole alkaloid, can be isolated on a large scale from the rhizomes of Glycosmis stenocarpa. This alkaloid has demonstrated various biological effects, such as anticancer, antiviral, and anti-inflammatory activities [12]. Furthermore, murrayafoline A is also reported to enhance contractility and increase Ca2+ currents [ICa1.2] mediated by the Cav1.2 channels in rat cardiomyocytes [13]. However, murrayafoline A is a non-polar and disadvantageous to the application in medicinal chemistry. An approach for enhancing this property is its hybridization with different bioactive compounds, including thiols such as 1,3,4-oxadiazole-2-thiol derivatives, amines, and phenols. Among these, the 1,3,4-oxadiazole-containing thiols can be selected for this purpose due to the biodiversity of bioactivities, including antiviral, cytotoxic, and antimicrobial properties [14]. As mentioned in our previous report, 6a-c and their analogues have been hybridized with zerumbone, yielding conjugates with prominent anticancer activity compared to zerumbone. Therefore, in this paper, new murrayafoline A derivatives with the thiols 6a-c have been synthesized and explored for their cytotoxic activities.
2. MATERIAL AND METHODS
2.1. Material
Murrayafoline A 1 was prepared from the rhizomes of Glycosmis stenocarpa the following procedure as described in our studies [11]. Other chemicals were purchased from Merck without further purification. Technical solvents were re-distilled using a Vigreux column before use. A Bruker Avance ̀ 600 MHz spectrometer was used for measuring proton and carbon spectra in DMSO-d6 at the Institute of Chemistry (VAST). Mass spectra were obtained from an Agilent LC/MSD Trap SL at the Institute of Forensic Science, Ministry of Public Security. Thin-layer chromatography (TLC) was used for reaction control, and the elution condition for column chromatography. Silica gel (230-400 mesh) was used for column chromatography.
2.2. Methods
Synthesis of the N-glycidyl-1-methoxy-3-methyl-9H-carbazole (N-glycidylmurrayafoline A) 2
N-glycidylmurrayafoline A 2 was synthesized using the procedure as described in reference [12].
Synthesis of hydrazide 5a-c
Each acid: 4-methoxyphenyl acetic acid 3a, pyridine 3-carboxylic acid 3b, and pyridine-2-carboxylic acid 3c was converted to its corresponding ester by refluxing in absolute ethanol with concentrated sulfuric acid as the catalyst. The resulting esters were subsequently treated with hydrazine hydrate in methanol, producing hydrazides 5a-c as white solids, which were used for the oxadiazole preparations 6a-c without purification.
Synthesis of the 1,3,4-oxadiazole-2-thiols 6a-c
Thiols 6a-c were synthesized according to the procedure of Hassan and colleagues [15] with minor modifications.
A mixture was prepared from one of the hydrazides (5 mmol), carbon disulfide (2.4 equiv.), and KOH (2.4 equiv.) in ethanol (25 mL), which was continuously refluxed on an oil bath for 14 hrs. The solvent was partially evaporated until 1/3 of the original volume remained. The mixture was dissolved in water, and then the pH was adjusted to 5 with glacial acetic acid, resulting in the formation of a solid. This solid was then collected, washed with water, and dried to receive the corresponding thiols 6a-c. Crude oxadiazoles 6a-c were purified by flash chromatography.
5-(4-methoxybenzyl)-1,3,4-oxadiazole-2-thiol (6a)
Yield 70%, white solid, m.p. 106-108 oC; 1H-NMR (600 MHz, DMSO-d6, δ (ppm), J (Hz)): 13.45 (s, 1H, -SH), 7.24 (d, J: 9.0, 2H, H2'', H6''), 6.92 (d, 9.0, 2H, H3'', H5''), 4.05 (s, 2H, 1''-CH2-), 3.74 (s, 3H, 4''-OCH3).
5-(pyridin-3-yl)-1,3,4-oxadiazole-2-thiol (6b)
Yield 60%, white solid, m.p. 229-230 °C; 1H-NMR (600 MHz, DMSO-d6, δ (ppm), J (Hz)): 14.77 (s, 1H, -SH), 9.05 (dd, J: 1.2, 2.4, 1H, H2''), 8.80 (dd, J: 1.2, 4.8, 1H, H6''), 8.26 (m, 1H, H4''), 7.63 (m, 1H, H5'').
5-(pyridin-2-yl)-1,3,4-oxadiazole-2-thiol (6c)
Yield 62%, white solid, m.p. 205-207 oC; 1H-NMR (600 MHz, DMSO-d6, δ (ppm), J (Hz)): 14.85 (s, 1H, -SH), 8.76 (dd, J: 1.2, 4.8, 1H, H6''), 8.03 (m, 2H, H5'', H4''), 7.64 (m, 1H, H3'').
General procedure for the synthesis of β-thioether alcohols 7a-c
A solution of 2 (267 mg, 1 mmol), some drops of triethylamine (TEA), and each 2-mercapto-1,3,4-oxadiazole consists of: (6a), (6b), and (6c) (1 mmol) in dry DMF (5 mL) was stirred overnight at 100oC until TLC (n-hexane: acetone 2:1 v/v) indicated the absence of 2. The solvent was removed by rotary evaporation. Then, cold water was added, and the resulting mixture was acidified with 1.5 M HCl to a pH of 4.5. It was then extracted with EtOAc (3×25 mL). The combined EtOAc layer was washed with water and then dried over anhydrous sodium sulfate. The solvent was removed by rotary evaporation, and the resulting residue was purified by column chromatography using a 2:1 n-hexane:acetone eluent to give 7a-c.
Cytotoxic activity
The in vitro anticancer evaluation of the β-thioether alcohols 7a-c against the tested cancer cell lines (A549 and Hep-2C) and non-cancer cell (Vero) was conducted at the Department of Experimental Biology, Institute of Chemistry, according to the described methods [16, 17].
3. RESULTS
The procedure for the synthesis of three new β-thioether alcohol derivatives 7a-c of murrayafoline A with 1,3,4-oxadiazole-2-thiol derivatives at the 5-position is divided into three stages as outlined in the following Scheme:
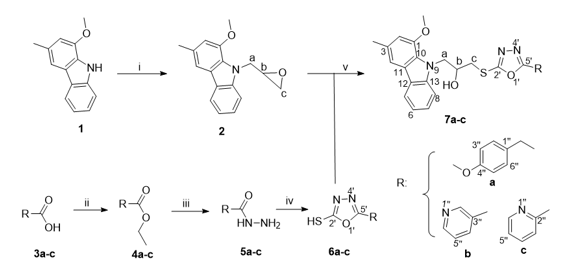
Reagents and conditions: i) Epichlorohydrin/NaH, THF, ii) EtOH, H2SO4, reflux, iii) NH2-NH2.H2O, MeOH, reflux, iv) CS2, KOH, EtOH, reflux, 12hrs, then CH3COOH, v) 2, TEA, DMF, 75oC, 24 hrs.
Scheme 1. The synthesis of β-thioether alcohols 7a-c
After purification using column chromatography, the three target derivatives 7a-c were structurally characterized by NMR and HRMS spectral data. All the detailed spectral data for structural interpretation of 7a-c were summarized as follows:
1-(1-methoxy-3-methyl-9H-carbazol-9-yl)-3-((5-(4-methoxybenzyl)-1,3,4-oxadiazol-2-yl)thio) propan-2-ol (7a).
Yield 80%, colorless oil; 1H-NMR (600 MHz, DMSO-d6, δ (ppm)), J (Hz)): 8.02 (d, J: 7.8, 1H, H5); 7.56 (d, J: 8.4, 1H, H8); 7.51 (s, 1H, H4); 7.36 (dd, J: 1.2, 7.8, 1H, H7); 7.15 (m, 3H, H2'', H6'', H6); 6.87 (dd, 3.8, 7.2, 2H, H3'', H5''); 6.82 (s, 1H, H2); 5.51 (d, 5.4, 1H, -OH); 4.71 (dd, J: 5.4, 14.4, 1H, Ha1); 4.47 (dd, J: 7.2, 14.4, 1H, Ha2); 4.23 (m, 1H, Hb); 4.07 (dd, 4.2, 16.2, 2H, 1''-CH2-1, 1''-CH2-2 ); 3.86 (s, 3H, 1-OCH3); 3.72 (s, 3H, 4''-OCH3); 3.37 (m,1H, Hc1); 3.29 ( dd, 7.2, 13.2, 1H, Hc2); 2.45 (s, 3H, 3-CH3); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 166.7 (C5'); 163. 7 (C2'); 158.4 (C4''); 146.1 (C1); 141.1 (C13); 129.9 (C2'', C6''); 128.7 (C3); 127.3 (C10); 126.0 (C1''); 125.3 (C7); 124.1 (C12); 122.1 (C11); 119.9 (C5); 118.6 (C6); 114.1 (C3'', C5''); 112.4 (C4); 110.0 (C8); 109.1 (C2); 69.1 (Cb); 55.5 (1-OCH3); 55.0 (4''-OCH3); 49.8 (Ca); 36.9 (Cc); 29.8 (1''-CH2); 21.3 (3-CH3). ESI-HRMS calculated for C27H27N3O4S: [M+H]+ (m/z): 490.1800, found: 490.1792.
1-(1-methoxy-3-methyl-9H-carbazol-9-yl)-3-((5-(pyridin-3-yl)-1,3,4-oxadiazol-2-yl)thio)propan-2-ol (7b).
Yield 82%, white solid, m.p. 58-59oC; 1H-NMR (600 MHz, DMSO-d6, δ (ppm)), J (Hz)): 9.05 (d, J: 1.8, 1H, H2''); 8.78 (dd, J: 1.8, 4.8, 1H, H6''); 8.18 (dd, J: 1.8, 7.8 , 1H, H4''); 8.02 (d, J: 7.8, 1H, H5); 7.60 (m, 2H, H8, H5''); 7.50 (s, 1H, H4); 7.38 (dt, J: 1.2, 8.4, 1H, H7); 7.13 (t, J: 7.5, 1H, H6); 6.84 (s, 1H, H2); 5.60 (J: 4.2, 1H, -OH); 4.77 (dd, J: 6.0, J: 14.4, 1H, Ha1); 4.55 (dd, J: 7.2, 14.4, 1H, Ha2); 4.32 (s, 1H, Hb); 3.93 (s, 1H, 1-OCH3); 3.48 (dd, J: 4.2, 13.2, 1H, Hc1); 3.41 (t, J: 7.2 Hz, 1H, Hc2); 2.44 (3-CH3); 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 164.7 (C5'); 163.1 (C2'); 152.3 (C2''); 146.9 (C6''); 146.1 (C1); 141.0 (C13); 133.8 (C4''); 128.8 (C3); 127.3 (C10); 125.3 (C7); 124.23 (C3''); 124.2 (C12); 122.1 (C11); 119.9 (C5); 119.6 (C5''); 118.6 (C6); 112.4 (C4); 110.0 (C8); 109.1 (C2); 69.2 (Cb); 55.6 (1-OCH3); 49.8 (Ca); 37.18 (Cc); 21.3 (3-CH3). ESI-HRMS calculated for C24H22N4O3S: [M+H]+ (m/z): 447.1491, found: 447.1485.
1-(1-methoxy-3-methyl-9H-carbazol-9-yl)-3-((5-(pyridin-2-yl)-1,3,4-oxadiazol-2-yl)thio) propan-2-ol (7c).
Yield 77%, white solid, m.p.150-151oC; 1H-NMR (600 MHz, DMSO-d6, δ (ppm)), J (Hz)): 8.74 (m, 1H, H6''); 8.03 (m, 3H, H5, H5'', H4''); 7.61 (m, 2H, H8, H3''); 7.51 (s, 1H, H4); 7.38 (dt, J: 1,2, 8.4, 1H, H7); 7.13 (dt, J: 0.6, 7.8, 1H, H6); 6.84 (s, 1H, H2); 5.60 (d, J: 6.0 Hz, 1H, -OH); 4,78 (dd, J: 4.8, 14.4, 1H, Ha1); 4.55 (dd, J: 7.2, 14.4, 1H, Ha2); 4.34 (m, 1H, Hb); 3.94 (s, 1H, 1-OCH3); 3.52 (dd, J: 6.0, 15.0, 1H, Hc1); 3.42 (dd, J: 7.8, 13.2, 1H, Hc2); 2.45 (s, 1H, 3-CH3). 13C-NMR (150 MHz, DMSO-d6, δ (ppm)): 165.1 (C5'); 164.1 (C2'); 150.2 (C2''); 146.1 (C1); 142.4 (C6''); 141.1 (C13); 137.8 (C4''); 128.7 (C3); 127.4 (C10); 126.3 (C3''); 125.3 (C7); 124.2 (C12); 122.6 (C5''); 122.1 (C11); 119.9 (C5); 118.6 (C6); 112.4 (C4); 110.0 (C8); 109.1 (C2); 69.13 (Cb); 55.57 (1-OCH3); 49.8 (Ca); 37.2 (Cc); 21.3 (3-CH3). ESI-HRMS calculated for C24H22N4O3S: [M+H]+ (m/z): 447.1491, found: 447.1483.
Spectra for the structural determination of the representative 7a were illustrated in Figure 2 and Figure 3 below.
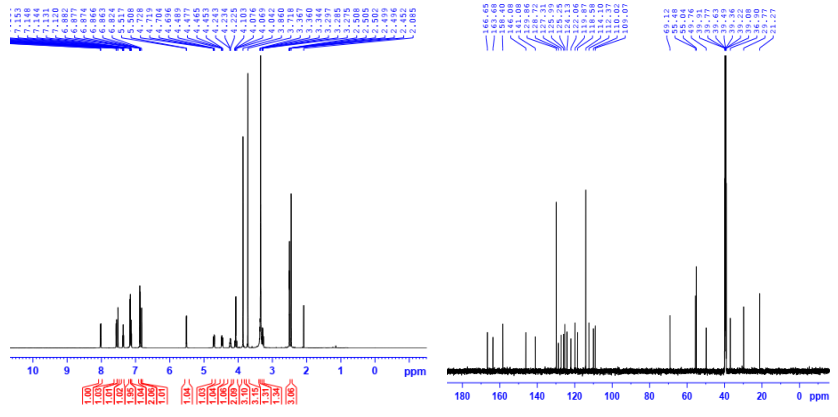
Figure 2. 1H-NMR and 13C-NMR spectra of the derivative 7a
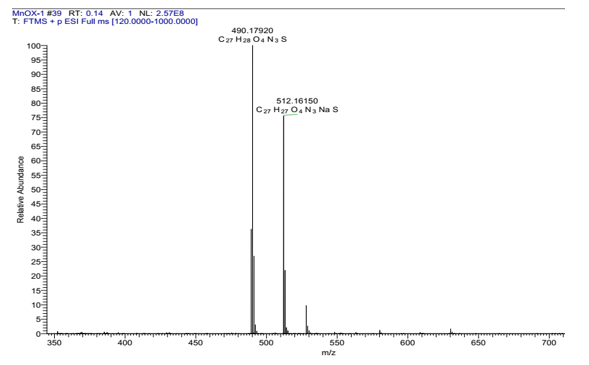
Figure 3. HRMS spectrum of the derivative 7a
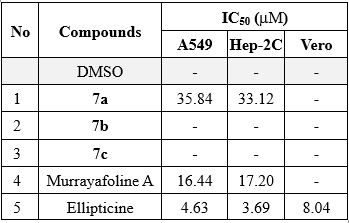
Finally, three synthesized derivatives 7a-c were screened for cytotoxic activity against human cancer A549, Hep-2C, and normal VERO cell lines. Compounds were reported to have no activity when the IC50 values were greater than 20 µg/mL. Their potencies were indicated in Table 1.
Table 1. Cytotoxic results of derivatives 7a-c
4. DISCUSSION
Both the -N9H group of murrayafoline A and the -SH group of mercapto compounds are nucleophilic agents; therefore, the connection between them can be completed by different linkers such as carboxyethyl, alkylene, and isopropanol. Among these linkers, the isopropanol linker is chosen because this structural group is an indispensable part of beta-blocker compounds. Such structural types of murrayafoline A derivatives and their cytotoxic activities have also been published by our study [12]. When the amine moieties are replaced by mercapto compounds, thioethers of beta-amino alcohols are produced. The synthesis of these compounds is divided into three stages as outlined in Scheme 1.
In the first stage, murrayafoline A 1 was N-alkylated with epichlorohydrin using a strong base, NaH, in THF to yield an intermediate, 1-methoxy-3-methyl-N-glycidylcarbazole 2 (65% yield), as described in our previous report [12]. In the second stage, oxadiazole thiols 6a-c were prepared from the corresponding acids 3a-c through a three-step procedure, which included esterification using ethanol and sulfuric acid as a catalyst to form esters 4a-c. These esters were then converted into the corresponding hydrazides 5a-c by nucleophilic substitution with hydrazine. In the final step, oxadiazole thiols 6a-c were obtained in 50-60% overall yields by cyclizing them with carbon disulfide and potassium hydroxide in ethanol. Although the synthesis and characterization of oxadiazoles 6a-c have been documented [18,19], their proton NMR spectra were still used to verify the structures. Their 1H-NMR data were consistent with the structures.
In the final stage, the opening of 1-methoxy-3-methyl-N-glycidylcarbazole 2 with thiols 6a-c in DMF, in a sealed glass reaction tube covered with a Teflon cap, and was performed at 100°C for 48 hours in the presence of TEA to produce 7a-c in yields ranging from 77% to 82%.
The structures of 7a-c were elucidated by ¹H-, ¹³C-NMR, and HRMS spectral data. The analysis of the signals in their proton and carbon spectra showed that the signals of the murrayafoline A skeleton were similar in both position and multiplicity, with a positional shift of <1ppm. The spectral differences between compounds 7a-c were only detected when comparing the signals of the mercapto moieties with each other. Interestingly, for compounds 7a-c, all the signals of the protons and carbons of the mercapto moiety were fully present, clear, and could be attributed in detail. Among the thioethers 7a-c, the compound 7a was selected for structural elucidation by the mentioned spectra. As shown in the proton spectrum of 7a (Figure 2), the murrayafoline A core was characterized by the following signals: a doublet appeared at 8.02 ppm (J: 8.1 Hz) belonging to H5, and another doublet gave rise to 7.56 ppm (J: 8.1 Hz) was attributed to H8. Two singlet signals were observed at 7.51 and 6.82 ppm that were assigned to H4 and H2, respectively. A doublet of triplet signal at 7.36 ppm was attributed to H7; the resonance of proton H6 gave a signal overlapped by the signals derived from protons H2'' and H6'' of thiol 6a. The methoxy and methyl groups were identified by two singlets, appearing at 3.86 and 2.45 ppm, respectively. In the thiol moiety, two protons of the methylene group were recognized by two doublets (J: 16.2 Hz) at 4.09 and 4.06 ppm. The singlet at 3.72 ppm was attributed to the 4''-OCH3 group. Next, the remaining protons H3'' and H5'' were identified by a dd signal at 6.87 ppm (3.8 and 8.1 Hz), respectively. Finally, the isopropanol linker was confirmed by a doublet of the hydroxy group (J: 5.0 Hz), and a multiplet appearing at 4.23 ppm was attributed to H-b. The resonance of four protons: Ha1, Ha2, Hc1, and Hc2 belonging to two methylene groups (CH2a, CH2b) were observed at 4.71 ppm (dd, J: 5.4 and 14.4, 1H, Ha1); 4.47 (dd, J: 7.2 and 14.4, 1H, Ha2); 4.23 (m, 1H, Hb); and 3.37 (m,1H, Hc1); 3.29 ( dd, J: 7.2 and 13.2, 1H, Hc2), respectively.
The assignment of signals in the 13C-NMR of the derivative 7a (Figure 2) was performed based on the reported carbon signal data of murrayafoline A, thiol 6a, and isopropanol moieties in the references [12,16,17]. The murrayafoline A skeleton was characterized by the following signals: 146.1 (C1); 141.1 (C13); 128.7 (C3); 127.3 (C10); 125.3 (C7); 124.1 (C12); 122.1 (C11); 119.9 (C5); 118.6 (C6); 112.4 (C4); 110.0 (C8); 109.1 (C2); 55,5 (1-OCH3), and 21.3 (3-CH3). The corresponding signals to the thiol 6a framework were readily recognized based on their characteristic chemical shifts: 166.7 (C5'); 163.7 (C2'); 158.4 (C4''); 129.9 (C2'' and C6''); 126.0 (C1''); 114.1 (C3'' and C5''); 55.0 (4''-OCH3); and 29.8 (1''-CH2-). Finally, the presence of the isopropanol linker was confirmed by the carbon signals at 69.1 (Cb), 49.8 (Ca), and 36.9 (Cc). Thus, the spectral data of 7a are consistent with its structure. The structures of 7b and 7c were also elucidated based on the spectral data of 7a and those reported in references.
The cytotoxic evaluations are listed in Table 1. The first observation on the cytotoxicity revealed that all three β-thioether alcohols, 7a-c, were non-toxic to the normal cell line VERO. Two pyridine-containing β-thioether alcohols in the oxadiazole moiety did not exhibit cytotoxic activity toward the two cancer cell lines tested. Among the three presented β-thioether alcohols, only derivative 7a displayed activity against two cell lines, Hep-2C and A549 (IC50 values: 33.12 and 35.84 µM, respectively). In comparison, its potency was weaker than that of parent compound murrayafoline A, which demonstrated cytotoxic activity against A549 and Hep-2C cell lines with IC50 values of 16.44 and 17.20 µM, respectively.
5. CONCLUSION
For the first time, three β-thioether alcohols of murrayafoline A bearing various 1,3,4-oxadiazole-2-thiol moieties are presented with a profile of their structures and cytotoxicity toward cancer cell lines, A549 and Hep-2C.
The preliminary evaluation of the thiol role in cytotoxic potential of the 7a-c revealed that the pyridine-containing derivatives of the thiols had no beneficial cytotoxic activity against cancer cell lines, while the 4-methoxybenzyl group of the thiol showed weak activity. Such a profile will serve as a valuable reference for future studies on this subject.
Acknowledgment: This research is funded by Grant from the Vietnam Academy of Science and Technology with the code NCVCC07.02/25-25.
Statement on the use of Generative AI: The authors affirm that no generative AI tools were used to create or modify the scientific content of this manuscript. All analyses, interpretations, and conclusions are entirely the work of the authors.
Author contributions: L.Y.Nhi: investigation, formal analysis; B.T.Chuyen: investigation; T.V.Anh: methodology, data curation; N.T.Anh, T.K.Vu: supervision, writing-review & Editing; T.N.Hung: methodology, formal analysis; N.M.Cuong: data curation, resource; L.V.Chinh: conceptualization, supervision, writing original draft.
Conflict of interest statement: The authors declare no conflict of interest.
References
2. M. A. Sukari, K. Ahmad, J. Haron, and R. Muse, Carbazole alkaloids from roots of Murraya koenigii, J. Analytical Sciences, vol. 7, pp. 263-265, 2001.
3. T. Sripisut, S. Laphookhieo, et al., Chemical constituents from the roots of Clausena excavata and their cytotoxicity, Rec. Nat. Prod., vol. 6, no. 4, pp. 386-389, 2012.
4. N. M. Cuong, T. Q. Hung, T. V. Sung, and W. C. Taylor, A new dimeric carbazole alkaloid from Glycosmis stenocarpa roots, Chem. Pharm. Bull., vol. 52, no. 10, pp. 1175-1178, 2004. DOI: 10.1248/cpb.52.1175
5. J. C. Kim, J. Wang, M.-J. Son, N. M. Cuong, and S.-H. Woo, Sensitization of cardiac Ca²⁺ release sites by protein kinase C signaling: evidence from action of murrayafoline A, Pflügers Arch., vol. 467, no. 7, pp. 1607-1621, 2015. DOI: 10.1007/s00424-014-1589-9
6. N. M. Cuong, H. Wilhelm, A. Porzel, N. Arnold, and L. Wessjohann, 1-O-Substituted derivatives of murrayafoline A and their antifungal properties, Nat. Prod. Res., vol. 22, no. 16, pp. 1428-1432, 2008. DOI: https://doi.org/10.1080/14786410802006033
7. G. Bringmann, A. Ledermann, and G. Francois, Dimeric murrayafoline A, a potential bis-carbazole alkaloid: biomimetic synthesis, atropoisomer separation, and antimalarial activity, Heterocycles, vol. 40, no. 1, pp. 293-300, 1995. DOI: 10.3987/COM-94-S30
8. S. Saponara, M. Durante, O. Spiga, P. Mugnai, G. Sgaragli, T. T. Huong, P. N. Khanh, N. T. Son, N. M. Cuong, and F. Fusi, Functional, electrophysiological and molecular docking analysis of the modulation of Cav1.2 channels in rat vascular myocytes by murrayafoline A, Br. J. Pharmacol., vol. 173, no. 2, pp. 292-304, 2016. DOI: https://doi.org/10.1111/bph.13369
9. D. P. Chakraborty and B. K. Chowdhury, Synthesis of murrayanine, J. Org. Chem., vol. 33, no. 3, pp. 1265-1268, 1968. DOI: https://doi.org/10.1021/jo01267a083
10. C. J. Moody and T. Martin, A new route to 1-oxygenated carbazoles. Synthesis of the carbazole alkaloids murrayafoline-A and murrayaquinone-A, J. Chem. Soc., Perkin Trans. 1, pp. 235-240, 1988. DOI: https://doi.org/10.1039/P19880000235
11. T. Q. Toan, P. T. Dan, N. T. Duong, L. V. Huyen, D. H. Nghi, L. X. Duy, N. P. Hung, N. M. Cuong, L. V. Chinh, P. M. Quan, V. T. Huyen, P. T. Nhut, and N. P. T. Dung, Optimization of Murrayafoline A ethanol extraction process from the root of Glycosmis stenocarpa, and evaluation of tumorigenesis inhibition activity on Hep-G2 cells, Open Chem., vol. 19, pp. 830-842, 2021. DOI: https://doi.org/10.1515/chem-2021-0067
12. L. V. Chinh, L. D. Anh, N. T. Nga, N. T. H. Ly, V. T. Ha, T. Q. Toan, N. M. Cuong, and T. K. Vu, Synthesis and in vitro cytotoxic evaluation of novel murrayafoline A derived β-amino alcohols, Lett. Org. Chem., vol. 14, no. 8, pp. 603-611, 2017. DOI: https://doi.org/10.2174/1570178614666170629131555
13. A. Hassan, S. Garpil, and I. Khan, Synthesis, characterization, and antifungal activity of 5-substituted 1,3,4-oxadiazole-2-thiols, Asian J. Chem., vol. 23, no. 5, pp. 2007-2010, 2011.
14. K. Likhitwitayawuid, C. K. Angerhofer, G. A. Cordell, J. M. Pezzuto, and N. Ruangrungsi, Cytotoxic and antimalarial bisbenzylisoquinoline alkaloids from Stephania evecta, J. Nat. Prod., vol. 56, no. 1, pp. 30-38, 1993. DOI: https://doi.org/10.1021/np50091a005
15. P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney, and M. R. Boyd, New colorimetric cytotoxicity assay for anticancer agents, J. Natl. Cancer Inst., vol. 82, pp. 1107-1112, 1990. DOI: https://doi.org/10.1093/jnci/82.13.1107
16. Q. R. Du, D. D. Li, Y. Z. Pi, J. R. Li, J. Sun, F. Fang, W. Q. Zhong, H. B. Gong, and H. L. Zhu, Novel 1,3,4-oxadiazole thioether derivatives targeting thymidylate synthase as dual anticancer/antimicrobial agents, Bioorg. Med. Chem., vol. 21, no. 8, pp. 2286-2297, 2013. DOI: https://doi.org/10.1016/j.bmc.2013.02.008
17. M. Hanif, K. Shoaib, M. Saleem, N. H. Rama, S. Zaib, and J. Iqbal, Synthesis, urease inhibition, antioxidant, antibacterial, and molecular docking studies of 1,3,4-oxadiazole derivatives, ISRN Pharmacol., vol. 2012, Article ID 928901, 9 pages. DOI: https://doi.org/10.5402/2012/928901